From Discussions VOL. 10 NO. 1Genome Profile of Ambystoma mexicanum
IN THIS ARTICLE
KEYWORDS
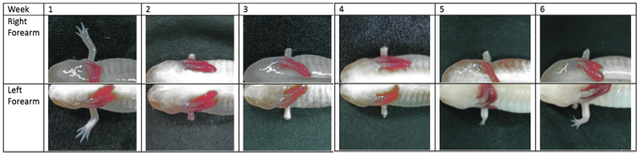
AbstractThe Ambystoma mexicanum, commonly known as the axolotl, possesses extraordinary regenerative abilities and is capable of reconstituting limbs, retina, liver, and even minor regions of the brain (Muneoka et al., 2008). At the most elementary level, regeneration is mediated by a cascade of epigenetic processes. With the aid of recent advances in Next Generation Sequencing (NGS), a study was conducted to identify and sequence the gene expression responsible for axolotl regeneration. Tissue samples from denervated limbs and regeneration blastema of 36 axolotls were sequenced by Illumina. Following de novo into 307,345 unigenes. Annotated sequences were analyzed against the Xenopus laevis database and sorted by gene oncology and clusters of orthologous groups were detected in the blastemal tissue and analyzed for gene expression; these assembled transcripts will pave the foundation for future research relating to wound regeneration, genomic stability, and cell differentiation. In addition, the results of this experiment provided evidence for the applicability of Illumina sequencing to yield comprehensive data for species lacking a reference genome. INTRODUCTIONThe axolotl has long been the model for studies on regeneration in organisms. But perhaps the most intriguing aspect of salamander regeneration is their cells' ability to "know" where on the proximal-distal axis the amputation took place and regenerate in the proper order (Ferris et al., 2010). Upon limb amputation, the epidermal cells migrate to cover the wound, which hardens into a layer of signaling cells called the apical epithelial cap (AEC), while truncated nerves innervating the limb release mitosis-stimulating factors, such as glial the blastema (Mchedlishvili et al., 2007). However, in the absence of an intact nerve supplxay at the site of amputation, these factors are not released and limb regeneration is terminated. At this stage, the regeneration blastema continues into their embryonic stages; these blastemal cells will eventually redifferentiate to form the mesodermal structures of a vertebrate limb. The Ambystoma mexicanum has a large genome composed of repetitive DNA components and functional elements (Smith et al., 2009). Recent studies have also shown the presence of Ambystoma-human non-redundant (nr) orthologous (Caron et al., 2001). In comparison to humans, whose genomic library has been approximated to be around 3.2 giga base pairs, the axolotl's genomic library is estimated to have around 10 times the amount of base pairs; current data suggests the axolotl has around 21.9 to 48 giga base pairs (Habermann et al., 2004). While the roles of these DNA components are not yet fully understood, it can be inferred that these DNA components harbor sequences that regulate the biological (Tamura et al., 2008). In the absence of a completed gene database, a comprehensive description of the spectrum of genes expressed during axolotl regeneration is unavailable. It is therefore inevitable that the sequencing of the axolotl genome is a foremost priority in understanding the cellular mechanisms by which this epimorphic process works. NGS technology provides a viable resource for sequencing the axolotl genome. NGS has not only enabled researchers to produce accurate results, but also to sequence DNA in a much shorter time (Parchman et al., 2010). In their work to recognize gene expression in regenerative tissues, Campbell and colleagues suggested the use of highly parallel 454 pyrosequencing to identify axolotl sequences (Campbell et al. that pyrosequencing of the axolotl genome produced over 1.7 million high-quality cDNA sequences without the use of a reference genome (Monaghan et al., 2012). An alternative to pyrosequencing is another NGS technique, clonal arrays that are then sequenced by reverse terminator bases (Bentley et al., 2008). Whereas Illumina boasts a larger coverage, higher reads per run, and more paired-end read, it also suffers from a shorter read length when compared to pyrosequencing. But for the purpose of sequencing an entire genome, Illumina offers a broader coverage of genes at a lower cost per run. Genetic data for salamanders is scarce, and the lack of a reference genome precludes the use of a referencebased assembly to acquire additional data. However, with the information obtained through NGS sequencing, de novo assembly can be utilized to generate a transcriptome composed of the mRNA sequence resources. Additional conclusions can be drawn through functional annotation to account for repeated sequences, while in situ hybridization can reveal the sites of gene expression. Through this method, a comparison between a regeneration blastema in levels of differentiated expressed genes. In conjunction with the hypothesis, an assumption was made that most of will be those relating to DNA regulation and synthesis. Therefore, the characterization of the axolotl transcriptome can clarify the functions in which these sequences are involved. METHODSAnimal Experimental ProceduresThe experimental sample size was comprised of 18 wild-types (WT) and 18 white mutants (d/d) reared from laboratory-farmed eggs. Each axolotl was kept separate in 14-20°C tap water with a pH level of 6.5-7.5. Anesthesia (0.1% MS-222) was used for all surgical procedures, and the experiment was done in accordance with the regulations of the Institutional Animal Care and Use Committee of the National Taiwan University College of Medicine. At a snout to vent length of 80 to 90 mm, the juvenile axolotls were denervated at the brachial plexus of the right supplies brachium. Amputation was performed on both forearms at the ulna one week later, and collection of tissue samples was conducted two weeks post-amputation (Figure 1) . One library was assembled from each of the acquired samples: blastemal cells from the left forearm and non-regenerating tissue from the right. Figure 1. Stages of regeneration in a (d/d) Amystoma mexicanum. The right forearm was denervated in week 1 followed by amputation on both forearms in week 2. Collection of tissues was conducted in week 4. Complete regeneration of the left forearm is observed in week 6.
Library PreparationRNA from both libraries was isolated and purified using Trizol Reagent and RNeasy Mini Kit, respectively. polymerase chain reaction (PCR) yielded second-stranded visible bands were eluted out of the gel using QlAquick Gel Extraction Kit and then amplified again by PCR. Illumina SequencingThe cDNA was sequenced on the Illumina GA IIx platform after the low-quality sequences had been removed from the libraries. The remaining fragments were assembled by three de novo transcriptome techniques: Trinity, transABySS, and SOAPdenovo (Robertson et al., 2010). Trinity and SOAPdenovo were run at the default k-mer settings, while trans-ABySS was run at k-mer values of 45, 46, and 89, as suggested by Robertson and his colleagues (2010). The assembled contigs were then sequenced again by Illumina, and contigs of at least 100bp in length were collected for functional annotation. Functional Annotation and Quantitative AnalysisContigs were input into Basic Local Alignment Search Tool (BLASTx) software. The data was assessed against the protein database of Xenopus laevis as a reference because it has a close evolutionary relationship with the axolotl (Altschul et al., 1997). The cut-off score for BLASTx analysis was set at an e-value of 1E-5. Contigs from each of the data sets were assigned a name and function to that of the matching protein in the Xenopus laevis genome based on the best BLASTx hit. If similar contigs were assigned to the same gene, the one with the lowest e-value was selected, and if these contigs shared the same e-value, the one with the longest base pairs was chosen. The non-redundant NCBI nucleotide database was also used for comparison. CLC Genomics Workbench 4.8 software was used for quantitative analysis with the parameter settings of 0.5 for the minimum length fraction, 0.95 for the minimum similarity fraction, and 10 for the maximum number of hits for a read. In situ HybridizationThe two genomic libraries were amplified by PCR using primers that contained the T7 (anti-sense) and SP6 (sense) promoters. Anti-sense and sense probes for the genes patched-2 and pax7 were synthesized with the primers and tagged with digoxigenin, an antibody-based marker. Blastemal tissues were sectioned at increments of 10μm, and alkaline phosphatase activities on the anti-DIG-antibodies were detected using a mixture of the BCIP/NBT solution.Continued on Next Page » From the Inquiries Journal Blog   Related ReadingJournalQuest is a free program to help academic student publications increase online readership and distribution. If you are interested in enrolling a journal at your school, please visit the JournalQuest website. Monthly Newsletter SignupThe newsletter highlights recent selections from the journal and useful tips from our blog. Suggested Reading from Inquiries Journal
Inquiries Journal provides undergraduate and graduate students around the world a platform for the wide dissemination of academic work over a range of core disciplines. Representing the work of students from hundreds of institutions around the globe, Inquiries Journal's large database of academic articles is completely free. Learn more | Blog | Submit Follow IJ
Latest in Biology |