The Importance of the Ras-Raf Pathway in Cancer Development
By
2016, Vol. 8 No. 08 | pg. 1/1
IN THIS ARTICLE
KEYWORDS
AbstractThe Ras/Raf pathway is a crucial cell signaling pathway utilized by eukaryotic cells for growth and proliferation, and it is highly conserved amongst all eukaryotic organisms. Mutations in this pathway lead to uncontrolled growth and proliferation of cancerous cells, effectively giving them a major advantage over normally functioning cells. Not surprisingly, numerous specific mutations and epigenetic changes to these proto-oncogenes contribute to carcinogenesis in colon, skin, breast, lung, and other cancers. In this review, I explore the role of the Ras/Raf pathway in many different types of cancer. I highlight up and coming gene therapy techniques that allow for targeting of the Ras/Raf pathway. The future is bright as treatment is finally reflecting the fact that cancer is different in everyone, and individualized treatment is realistic and effective. IntroductionCancer is one of the most complex and intriguing illnesses studied by the medical and scientific community. It is particularly distinctive due to the fact that development of cancer can be unique in each patient. Factors influencing the advancement of cancer include a seemingly limitless pool of modalities and influential factors constituted by genes and the environment. These factors cannot be considered separate from one another; rather the interactions between a person’s genetic background and environment can combine in a wide variety of ways. The places we live, the air we breathe, the food we consume, the amount of physical activity we subject ourselves to, and many other factors influence our susceptibility for cancer development. Even though many different genetic aspects can influence cancer development, some are utilized more so than others. For example, many different types of cancer will manipulate the Ras/Raf or MAPK pathway for growth advantages partly due to the crosstalk with other fundamental pathways. The Ras/Raf pathway is fundamental to every eukaryotic cell’s ability to grow, proliferate, and even undergo programmed cell death. (1)Yet, this primeval and conserved pathway is still not completely understood; proteins involved in the Ras/Raf pathway are still being discovered, particularly in light of biological cross talk that occurs in this pathway.To initiate the Ras/Raf signaling cascade, a ligand, such as EGF (epidermal growth factor), first binds to its cognate receptor, such as EGFR (epidermal growth factor receptor). This in turn allows for the phosphorylation of the cytoplasmic portion of the receptor. Next, the protein GRB2 binds to the EGFR via its SH2 domain, and undergoes a conformational change which allows SHC and SOS to bind to its SH3 domain. This complex activates SOS, which results in SOS phosphorylating Ras-GDP (also known as the inactive form of the protein Ras) so that it may become activated and form Ras-GTP; thus begins the kinase cascade portion of this growth and proliferation pathway. Activation of Ras causes the phosphorylation of the downstream protein known as Raf. (2) Raf’s kinase activity also becomes activated, and it in turn phosphorylates a protein known as MEK. Activated MEK phosphorylates and activates the protein kinase ERK. ERK travels from the cytoplasm into the nucleus where it carries out a variety of functions. ERK regulates levels of expression of other proteins such as ELK, JUN, or YAN as well as synchronizing activity of transcription factors controlling a number of genes related to the cell cycle, growth, proliferation, differentiation, and apoptosis. (Figure 1) Figure 1- The EGFR pathway (3) 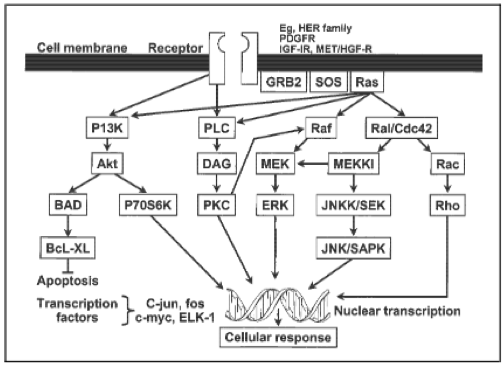 The Ras/Raf Pathway in Cancer Pathogenesis and TumorigenesisCancer is a complex and diverse disease that can affect essentially all types of cells and tissues. Tumorigenesis, though seemingly random, is based on a number of events related to inherent genetic factors as well as environmental factors resulting in genetic or cellular turnover changes. Though the “two-hit mechanism” is a common explanation to tumorigenesis, it may take up to six or seven independent events to induce neoplasia. This explains why many cancers take decades to develop, even if a person has familial or de novo genetic susceptibility to a specific type of cancer when they are born. However, as the dysplastic cells gain access to more advantageous mutations as time goes by, the last few genetic hits can occur more quickly than the primary advantageous mutations. This is due to genetic instability in dysplastic cells, resulting in the probability of development of other mutations (which may not necessarily be beneficial) to increase. Manipulation of the Ras/Raf pathway is common in many types of cancer because upregulating or downregulating specific proteins in the pathway can often result in significant advantages. For example, upregulating the activated form of Ras, which controls not only the Ras/Raf pathway but also the P13K and RalGDS pathways, can result in increased calcium signaling, proliferation, growth, and transformation as well as detrimentally affecting cell adhesion and apoptosis. (2) This will allow the aberrant cells to flourish at the expense of healthy, normally functioning cells. A detailed description of how Ras family members contribute to cancer follows. Mutations to the Ras/Raf and other closely related pathways are often mutually exclusive given that a single hit to the pathway is enough to cause atypical effects such as upregulation of cell proliferation or downregulation of apoptosis. Therefore, another mutation to the pathway is not necessary and could result in a disadvantage for the dysplastic or neoplastic cells. (2) Though the Ras family proteins are popular targets for cancer formation, many other aspects of the Ras/Raf pathway are often mutated as well. The EGF receptor can be upregulated to increase the number of receptors on a cell’s surface, or point mutations can improve its affinity for ligands, which essentially keeps the receptor constantly active. Mutation of Raf is also relatively common in human cancers; as many as 40 different mutations in BRaf can contribute to neoplasia. (2) Mutations in the kinase cascade or proteins that permit crosstalk between the Ras/Raf pathway and the closely related P13K pathway make it difficult to target one protein of the pathway for therapy. Compensatory loops are often created which allow the neoplastic cells to avoid inhibition by a drug or therapy that targets one protein. Instead, a combination of inhibitors of both the Ras/Raf and P13K are currently being investigated as a superior strategy. (2) The Ras Gene FamilyActivating Mutations and Polymorphisms in CancerOne of the most commonly affected genes is the proto-oncogene Ras: upregulation of Ras is so advantageous that approximately 30% of tumors carry a Ras mutation. (4, 5) Ras generally rotates from its active GTP-bound form to its inactive-GDP form. Upregulating Ras by epigenetic events such as hypomethylation, or activating mutations that cause the Ras to be constantly bound to GTP result in upregulation of the remainder of the pathway. (6) This leads to uncontrollable cell proliferation, growth, and transformation due to upregulated transcription factors allowing constant transcription of genes influencing these cellular activities. The Ras protein family includes KRas, NRas, and HRas. Though each provide essentially the same function, amino acid sequences vary slightly as well as conformation of the protein. Genetic and epigenetic alterations to the Ras proto-oncogene are frequented found in pancreatic, lung, colon, breast, and melanoma cancers. (2, 7) Though NRas, and less frequently HRas, are sometimes targeted, KRas is generally the main target for upregulation in human cancers. Mutations in KRas make up 85% of total Ras mutations in concordance with cancer. NRas mutations appear less frequently at approximately 15% in cancer, and HRas less than 1%. (2) Activating point mutations of the KRas protein are very common in pancreatic and colon cancers, and also present less frequently in lung and breast cancers. KRas mutations are common in DNA mismatch repair (MMR) proficient cancers; hence why about 35% of colon cancers have a mutation in KRas. Mutations in DNA mismatch repair provide a steady stream of point mutations. This influx of point mutations and high turnover rate of colon epithelial cells, makes the possibility of mutations to Ras much more likely in colon cancers. (2) One study by Rajogopalan et. al found 169 mutations in KRas which included a variety of alterations to codons 12, 13, 59, and 61. (7) Through analysis of a number of different tumors, the authors also found that KRas mutations seem to occur after the initiation phase of tumorigenesis, but before the neoplastic population becomes malignant. Correspondingly, mutations to NRas also occur slightly earlier during tumorigenesis. NRas is a commonly mutated in metastatic melanoma cancers, about 19-28% of all cases, though it can also be found in a minute subset of colon and breast cancers. (2) In metastatic melanoma cases, an activating mutation to NRas at the location of its 61st codon appears most commonly in the primary lesion. NRas activation mutations occur early in development of metastatic melanomas—namely in the benign, primary lesions which precede malignancy. However, not only is the codon 61 activating mutation of NRas seen in primary lesions, but can be detected in last stages of malignancy. Using a wide variety of metastatic melanoma tumors as well as analyzing primary and later stage metastatic tumors which originated from the primary lesion, Omholt et. al found mutations appeared first in primary tumors rather than metastases. (8) These observations are suggestive of the fact that NRas mutations can occur in a relatively wide range of subsets to tumorigenesis, and that NRas mutations are maintained into later stages of development. (9) According to DeLuca et. al, mutations to HRas are found only in about 1% of breast cancers. Findings by Kompier et. al showed that HRas mutations occurred in 14 of 257 (less than 1%) samples of biopsies of primary tumors from patients with bladder cancer. (10) Though mutations to HRas seem to occur in the minority of bladder and breast cancers, polymorphisms of the HRas proto-oncogene are more relevant to cancer development than mutations occurring to HRas itself. In one GWAS study, rare HRas alleles associated with hormone receptor negative tumors have been connected to more aggressive breast cancer tumors, and more common in African-American females.(9) Garett et. al sampled a wide variety of women of different races and ages to determine a connection between polymorphism of the HRas allele and breast cancer. The study found that African-American women were 3-11 times more likely to develop aggressive breast cancer than the control groups, while women of European descent were only approximately 2 times more likely to develop aggressive breast cancer as compared to the control groups of the study. The same rare HRas polymorphism was found to be connected more frequently with breast cancer in younger women. The breast cancers in this subset of patients was commonly more aggressive than breast cancers sampled from older women. Early onset of breast cancer was linked to estrogen and progesterone receptor negative tumors, which is often linked to a worse prognosis and advanced-stage tumors. These ER and PR negative tumors were often linked with the rare HRas allele. The Impact of Activating Mutations and Epigenetic EventsThough activating mutations of Ras are often utilized by breast, metastatic melanoma, and colon cancers, activating mutations to the Raf gene family are perhaps the second or third most common event exploited by cancers. Similar to other proteins of the Ras/Raf pathway, the Raf family of proteins is a group of serine/threonine-specific kinases: ARaf, BRaf, and Raf-1 (also known as CRaf). The differences between each protein lie in seemingly infinitesimal differences in amino acid sequence, which in turn causes slight differences in conformation of each type of Raf protein. Disimilar to the Ras family, only one of the Raf family members potently promotes cancer development—BRaf. This is likely due to the fact that ARaf and CRaf have a much lower basal kinase activity than BRaf: BRaf phosphorylates the next kinase in the Ras/Raf cascade, MEK, more frequently than its other family members. More frequent basal kinase activity in addition to an activating mutations that drives kinase activity of Raf even higher, is most advantageous to aberrant cells. Not surprisingly, BRaf mutations are found in approximately 8-15% of human cancers, while CRaf mutations are found in less than 1%. (2, 11, 12) BRaf mutations are most common in thyroid, melanoma, colon, and breast cancer. Activating mutations to BRaf occur with a frequency over 40% in thyroid and melanoma cancers, approximately 15% of colon cancers, and 2% of breast cancers. (2) Concurrency with Ras mutations does not occur due to the fact that upregulating the Ras/Raf pathway via increasing the activity of one protein in the cascade is sufficient to create a sufficient gain of function. (11) BRaf activating mutations are largely point mutations to exons 11 and 15. (13) It has recently been discovered that point/missense mutations are not the only mechanism by which upregulation can be achieved. Interestingly enough, BRaf can be upregulated indirectly by epigenetic events. Hypermethylation of DNA repair genes such as hMLH1 (a gene involved in mismatch repair) can cause genomic instability sufficient to cause activation mutations in BRaf. This event commonly causes the activation mutations in exons 11 and 15 in BRaf. Hypomethylation of a CpG island or gene promoter region can cause overexpression of a target gene. Therefore, CpG island hypomethylation can cause effective overexpression of BRaf. (3) Targeting the Ras/Raf Pathway in Cancer TherapyThe scientific community has accepted that a cure-all for cancer most likely does not exist. Samples collected from cancer patients exhibit vastly different genetic profiles, and patients themselves have different environmental exposures and socioeconomic backgrounds. Almost every case of cancer is different, though some may have overlapping mutations or epigenetic events. Based on this, researchers have begun to look at individual gene therapies based on the genetic makeup of each patient’s cancer. Owing to the fact that the Ras/Raf pathway is often mutated, extensive research into targeting this pathway for cancer therapy is well underway. Cancer therapy targeting the Ras/Raf pathway ranges from researching small molecules for inhibiting mitogen-activated kinases, to antisense oligonucleotides. (14) Farnesyltransferase inhibitors are utilized to target Ras specifically because the Ras family of proteins have a unique farnesyl isoprenoid group which functions to achieve the correct conformation and localization. (14) Inhibiting the farnesyl pyrophosphate group or binding of the farnesyltransferase prevents the correct localization of Ras to the plasma membrane of the cell, which is necessary for the protein’s proper function—essentially Ras is no longer active. Therapeutic inhibitors of the farnesyl group or the farnesyltransferase can either directly inhibit or compete with the farnesyl pyrophosphate group of the Ras inhibitor. Though these farnesyltransferase inhibitors showed promise in murine models, FTIs aren’t completely effective because KRas and NRas can also be modified by another transferase known as gamma-glutamyltransferase, or GGT. (14, 15) A more promising therapy involves targeting HRAS or Raf-1 using antisense oligonucleotides, or ASOs. These oligonucleotides are synthetic, and are made to bind to specific regions of mRNA made by the HRas and Raf-1 genes prior to being translated to proteins. The antisense nucleotides bind, and inhibit protein production by either promoting degradation of the mRNA or by shutting down translation completely by not allowing transcriptional machinery to bind to the mRNA. (14) Raf-1 antisense oligonucleotide-treated mice had a significant reduction in the number of metastases while not affecting liver cell proliferation. (16) However, some problems with tumor uptake of the antisense oligonucleotides as well as nonspecific toxicity have stalled this therapy in its clinical trials. (14) One study found disease stabilization by two out of thrity-four patients, and no tumor regression even though toxic effects were so limited. However, levels of CA-125, a potential marker of cancer, decreased by 97% in a patient with ovarian cancer. Therefore, taking ASOs into phase II and phase III trials may be quite beneficial for cancer therapies. (17) Small molecule kinase inhibitors targeting upstream and downstream of Ras are showing promising development as well. Specific drugs like imatinib, an inhibitor of BCR-ACL, have the ability to target specific kinase-activating proteins in the pathway like Ras or MEK; by doing so, the proliferation, survival, and growth advantages cancers access, are effectively shut down. (14) MEK inhibitors seem to have a great specificity for their targets, and have a good reputation in safety. Table One has a list of MEK inhibitors and important details regarding each. Sorafenib, Raf kinase and VEGFR inhibitor, had significant activity in renal, colon, pancreatic, lung, and ovarian tumors. (18) 70% of patients with tumors taking Sorafenib, had shrinkage in size or stabilization. (19) However, many times, a kinase in the Ras/Raf pathway often has more than one target, so avoiding one protein isn’t necessarily detrimental. Therefore, small molecules will likely need to be combined with other cancer target therapies or cytotoxic agents. Table 1
Synopsis and DiscussionThe Ras/Raf pathway is so useful for cancer cells because of its influence on cell survival including regulation of apoptosis, cell cycling, proliferation, growth, cell motility, and angiongenesis. Therapies and cancer drugs which target the Ras/Raf pathway are still largely being developed today. Most successful therapies are still in the early stages of clinical trials. Kinase inhibitors have shown some of the most promising advances with the least major side-effects. Endnotes1. Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochemical Journal. 2000;351(2):289-305. 2. De Luca A, Maiello MR, D'Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert opinion on therapeutic targets. 2012;16(sup2):S17-S27. 3. Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. Journal of the National Cancer Institute. 2001;93(14):1062-74. 4. Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111-6. 5. Khosravi-Far R, Der CJ. The Ras signal transduction pathway. Cancer and Metastasis Reviews. 1994;13(1):67-89. 6. Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, et al. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Molecular and cellular biology. 1996;16(7):3923-33. 7. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934-. 8. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clinical Cancer Research. 2003;9(17):6483-8. 9. Garrett PA, Hulka BS, Kim YL, Farber RA. HRAS protooncogene polymorphism and breast cancer. Cancer Epidemiology Biomarkers & Prevention. 1993;2(2):131-8. 10. Kompier LC, Lurkin I, van der Aa MN, van Rhijn BW, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PloS one. 2010;5(11):e13821. 11. Emuss V, Garnett M, Mason C, Marais R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer research. 2005;65(21):9719-26. 12. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949-54. 13. Kambara T, Simms L, Whitehall V, Spring K, Wynter C, Walsh M, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53(8):1137-44. 14. Downward J. Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer. 2003;3(1):11-22. 15. Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070-4. 16. Khatib A-M, Fallavollita L, Wancewicz EV, Monia BP, Brodt P. Inhibition of hepatic endothelial E-selectin expression by C-raf antisense oligonucleotides blocks colorectal carcinoma liver metastasis. Cancer Research. 2002;62(19):5393-8. 17. Cunningham CC, Holmlund JT, Schiller JH, Geary RS, Kwoh TJ, Dorr A, et al. A phase I trial of c-Raf kinase antisense oligonucleotide ISIS 5132 administered as a continuous intravenous infusion in patients with advanced cancer. Clinical Cancer Research. 2000;6(5):1626-31. 18. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. Journal of Pharmacology and Experimental Therapeutics. 2005;315(3):971-9. 19. Ratain M, Flaherty K, Stadler W, O'Dwyer P, Kaye S, Xiong H, et al., editors. Preliminary antitumor activity of BAY 43-9006 in metastatic renal cell carcinoma and other advanced refractory solid tumors in a phase II randomized discontinuation trial (RDT). ASCO Annual Meeting Proceedings; 2004. 
From the Inquiries Journal Blog   Monthly Newsletter SignupThe newsletter highlights recent selections from the journal and useful tips from our blog. Suggested Reading from Inquiries Journal
Inquiries Journal provides undergraduate and graduate students around the world a platform for the wide dissemination of academic work over a range of core disciplines. Representing the work of students from hundreds of institutions around the globe, Inquiries Journal's large database of academic articles is completely free. Learn more | Blog | Submit Follow IJ
Latest in Biology |