Huntington’s disease is a progressive neurodegenerative disorder that affects around five people in every 100,000. It is caused by an increase in a polyglutamine region of the Huntingtin protein, resulting in a toxic gain of function mutation. However, the exact mechanism of pathogenesis, or even the exact role of the normal Huntingtin protein are not comprehensively understood. It is known that Huntingtin is ubiquitously expressed and has interactions with many different cellular components. Mutant Huntingtin has been extensively studied and its effects on cells noted but questions regarding its toxicity remain to be answered. Much research has been directed towards finding treatments for the condition and while some show progress, the scientific community is still some way away from a definitive therapy to slow or prevent the disease. This review will critically assess research that has been conducted into Huntington’s disease and propose possible future work.
Huntington’s disease (HD) is an autosomal dominant repeat disorder with complete penetrance and prevalence of around 4-8 per 100,000 (Harper, 1992). It is an expansion disorder, caused by an increase in the number of CAG (glutamine) repeats in the coding region of the gene for Huntingtin (Htt) protein on chromosome 4p16.3. This large polyglutamine (polyQ) expanse results in the production of mutant Huntingtin (muHtt), in a gain of function dynamic mutation (Landles & Bates, 2004). This new protein is pathogenic and causes neurodegeneration, leading to cognitive decline and progressive chorea.
The condition was first described by George Huntington (Huntington, 1872). He noticed that there was a high prevalence of the then unexplained condition amongst certain families- up to this point, sufferers had been thought to be possessed due to the chorea. In 1911, further research by Charles Davenport confirmed the genetic link and showed it was an autosomal dominant disorder (Davenport, 1911). A major breakthrough came in 1993 when the gene responsible was located at 4p16.3 (Huntington's Disease Collaborative Research Project, 1993). This paper showed where the mutation occurred, providing an insight into how the disease manifested in patients.
Non-Huntington’s sufferers have 36 or fewer repeats of CAG in their Htt gene, resulting in the production of normal cytoplasmic Htt (Strachan & Read, 2011). Once the polyQ region exceeds 36 repeats, it becomes unstable and the protein produced has different properties. Generally, a repeat length of between 36-39 will result in a delayed onset form of the disease, with less severe symptoms and a slower progression. Repeats of 40 or more CAGs are associated with almost complete penetrance by the age of 65 years (Langbehn, Brinkman, Falush, Paulsen, & Hayden, 2004).
Huntington’s disease is characterised by both motor and cognitive disorders of varying severity, which usually onset between the ages of 30 and 50 (Roos, 2010). The disease progresses for around 17-20 years and is eventually fatal. The most common cause of death due to Huntington’s is pneumonia, followed by cardiovascular disease (Heemskerk & Roos, 2012).
Motor symptoms include progressive chorea- involuntary, jerky movements. These initially begin in the extremities and gradually progress throughout the body, occurring when the sufferer is awake (Roos, 2010). The most common choreatic movement involves extension of the lower back muscles. As well as the chorea, the neuronal decline causes a loss of balance, leading to unstable walking. Sufferers are also affected by bradykinesia and akinesia. Dystonia is one of the first presenting symptoms and results in an abnormal posture presenting. Ultimately, these symptoms increase in severity and in the end stages of the disease patients struggle to stand and walk, resulting in them needing increasing levels of care.
Psychiatric disorders onset very early in the disease, usually before motor symptoms manifest. Depression is the most common, followed by guilt and anxiety (Julien, et al., 2007). Because of how early these symptoms appear (up to 20 years before motor symptoms in some cases, (Rosenblatt, 2007)) and the strong similarities in psychiatric symptoms in families (Lovestone, Hodgson, Sham, Differ, & Levy, 1996), it was initially unclear as to whether they were attributed to physical changes in the brain or simply genetics, somehow related to being at risk of the disease but with no neurodegeneration. A study by Julien et al. (2007) investigated this and found that lifetime psychiatric symptom presentation was no different in those with or without the mutation. The study did however find an increased incidence of depression amongst HD sufferers. Therefore, while psychiatric disturbances seem to have no relation to carrying the mutant gene, the HD gene confers an above average risk of depression.
Cognitive decline is a final major progressive symptom. This too can be present before motor signs and varies in its severity regardless of the overall progression of the disease. The decline is in ‘executive functions’, which are required to distinguish between relevant and useless information and to be able to organise and plan the patient’s life. Speech is generally unaffected, as is semantic memory, but other memory functions are gradually lost.
Unexplained weight loss is present in all patients from an early period. While it was first assumed this was due to the chorea, this has been shown to be untrue (Roos, 2010). It is more likely that the weight loss is due to the neuronal changes resulting in decreased appetite, slower functioning and difficulty with preparing and eating food. Recent research has shown that the longer the CAG repeat length, the faster the rate of weight loss in a patient (Aziz, et al., 2008) and has also associated hypothalamic neuronal loss and the subsequent metabolic and endocrine changes with a loss in body mass (Aziz, Swaab, Pjil, & Roos, 2007).
A final, secondary symptom is circadian rhythm disturbances. Human patients show disrupted sleep patterns, which have knock-on effects on their wellbeing. Mouse models of the disease show a progressive shift from nocturnal to daytime activity, eventually leading to a complete loss of circadian rhythm (Morton, et al., 2005). This symptom is due to a morphological change in the suprachiasmatic nucleus of the hypothalamus (responsible for maintaining circadian rhythm); specifically, a large reduction in both vasoactive intestinal polypeptide neurons (85%) and a reduction in arginine vasopressin neurons (33%) (van Wamelen, et al., 2013). These are key circadian rhythm neurotransmitters and mRNA levels for them were unchanged, showing the problem lies with their release, not synthesis.
A second, less common form of the condition is juvenile onset, defined as presenting before the age of 20. This accounts for ~10% of cases and is associated with a much larger than average repeat length (Walker, 2007). The key symptomatic difference commonly involves an absence of chorea. Instead, the sufferers exhibit rigidity. The disease also progresses much more quickly and is associated with a high incidence of seizures (Walker, 2007). Because this condition can begin from as early as two years and repeat length increases through generations, children of parents with the disease may display symptoms before them.
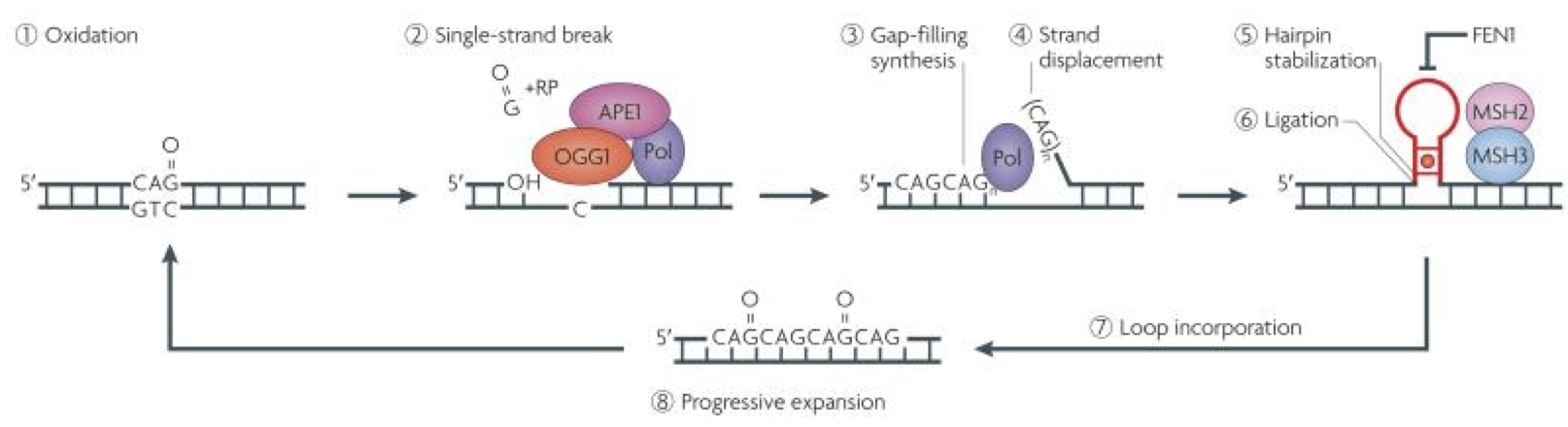
It is thought that the repeat sequence becomes elongated during DNA replication. Lagging strands of DNA are synthesised as a number of overlapping Okazaki fragments. Because of this, a specialised enzyme called FEN1 removes the overhang, allowing the fragments to correctly anneal. A mutated enzyme could join overlapping fragments, leading to the repeat expansion (Strachan & Read, 2011). Other research suggests that instead of occurring during DNA replication, it is the base excision repair system, involved in fixing single strand breaks due to oxidative damage, that causes expansion. The BER system can generate single strand overlaps which may then form repeats (Fig. 1) (McMurray, 2010). In human HD fibroblasts, which usually show no expansion when cultured in vitro, expansion can be induced by peroxide exposure, which damages DNA by oxidation (Kovtun, et al., 2007). This study also showed an in vivo escalating oxidation/excision system, which explains the age-dependant expansion seen through generations of sufferers of HD.
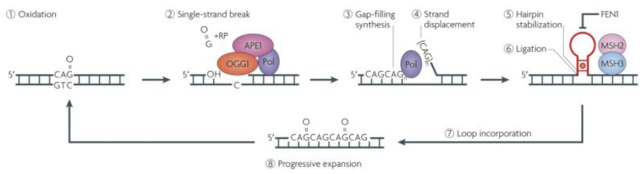 Figure 1. Showing the BER system. Oxidised, damaged DNA (1) is targeted by the BER enzyme complex (2), excising a single base pair gap which is then lengthened. The new strand is then synthesised (3). As shown in (4), strand displacement occurs during this process, forming a flap. This flap cannot be removed by FEN1 as commonly happens due to the stabilisation of the flap into a trinucleotide repeat hairpin (5). As shown by the red dot in the hairpin, A-A mismatches further stabilise the structure as the mismatch repair enzymes MSH2 and 3 bind. This hairpin may then be incorporated into the DNA (7) and this repeat process eventually leads to a ‘toxic oxidation cycle’ (8), where the repeat sequence progressively expands. Figure from (McMurray, 2010).
Figure 1. Showing the BER system. Oxidised, damaged DNA (1) is targeted by the BER enzyme complex (2), excising a single base pair gap which is then lengthened. The new strand is then synthesised (3). As shown in (4), strand displacement occurs during this process, forming a flap. This flap cannot be removed by FEN1 as commonly happens due to the stabilisation of the flap into a trinucleotide repeat hairpin (5). As shown by the red dot in the hairpin, A-A mismatches further stabilise the structure as the mismatch repair enzymes MSH2 and 3 bind. This hairpin may then be incorporated into the DNA (7) and this repeat process eventually leads to a ‘toxic oxidation cycle’ (8), where the repeat sequence progressively expands. Figure from (McMurray, 2010).
A final mechanism lies in the nucleotide excision repair system, consisting of two subsystems; global genome repair (GGR) and transcription-coupled repair (TCR). GGR is unlikely to be involved, as loss of the system in knockout mice has little effect on HD progression (Dragileva, et al., 2009). TCR involvement is more likely, as TCR knockout mice show a slower progression of contraction within repeat length (contractions are associated with a more instable repeat) (Lin & Wilson, Trascription induced CAG repeat contraction in human cells is mediated in part by transcription coupled nucleotide excision repair, 2007). However, the loops formed in this system are too small to form repeat sequences, meaning it is more likely that the BER system is involved in the CAG repeat expansion seen in HD.
A child of a heterozygous sufferer of HD has a 50% chance of inheriting the disease. HD was the first condition for which premanifest diagnosis information could be obtained, allowing for genetic counselling of potential sufferers and families expecting children. For genetic counselling, as long as the applicant meets none of the defined exclusion criteria, an initial consultation followed by a blood test 4-6 weeks later is available (International Huntington Association, 1994). For prenatal diagnosis, chorionic villi sampling can take place between the 10th and 12th week of pregnancy, as long as the parents are aware of their own genetic status. More recently, in vitro fertilisation has become involved. At the eight cell stage, one cell is removed and tested for HD and only embryos that do not have the disease will be implanted (Roos, 2010). Genetic testing is relatively simple, as the only diagnostic marker analysed is the length of the CAG repeat in the Htt gene.
Interestingly, despite the efficacy of the test, fewer than 5% of at risk individuals are actually tested (Walker, 2007). This is attributed to the lack of an effective treatment- those who undergo testing are generally making family or career choices. Around 1% of those who are diagnosed as sufferers attempt suicide (Almqvist, Bloch, Brinkman, & Crauford, 1999), meaning the exclusion criteria must be rigorously examined before testing and those who test positively must be adequately educated to prevent them being misled about the disease.
Huntington’s disease shows a genetic phenomenon called anticipation, a process where the age of onset in children is earlier than that of the parents (Strachan & Read, 2011). This earlier onset is usually accompanied by an increase in the severity of symptoms. A study by Ridley et al. (1988) found that generally the change in age of onset is equal regardless of whether the disease was inherited from the mother or father but in a small proportion of paternal inheritance cases (6%), the age of onset is lowered by around 20 years. This specific pedigree was associated with early motor rather than psychiatric symptoms. They further postulated that the reason for the small number of sufferers of this subset was due to the fact that constantly reducing the age of onset would eventually lead to such an early disease progression that the sufferer would be unable to reproduce, preventing the line continuing. Anticipation means it is essential that children of sufferers of the disease are tested as early as possible, in order to begin counselling and treatments.
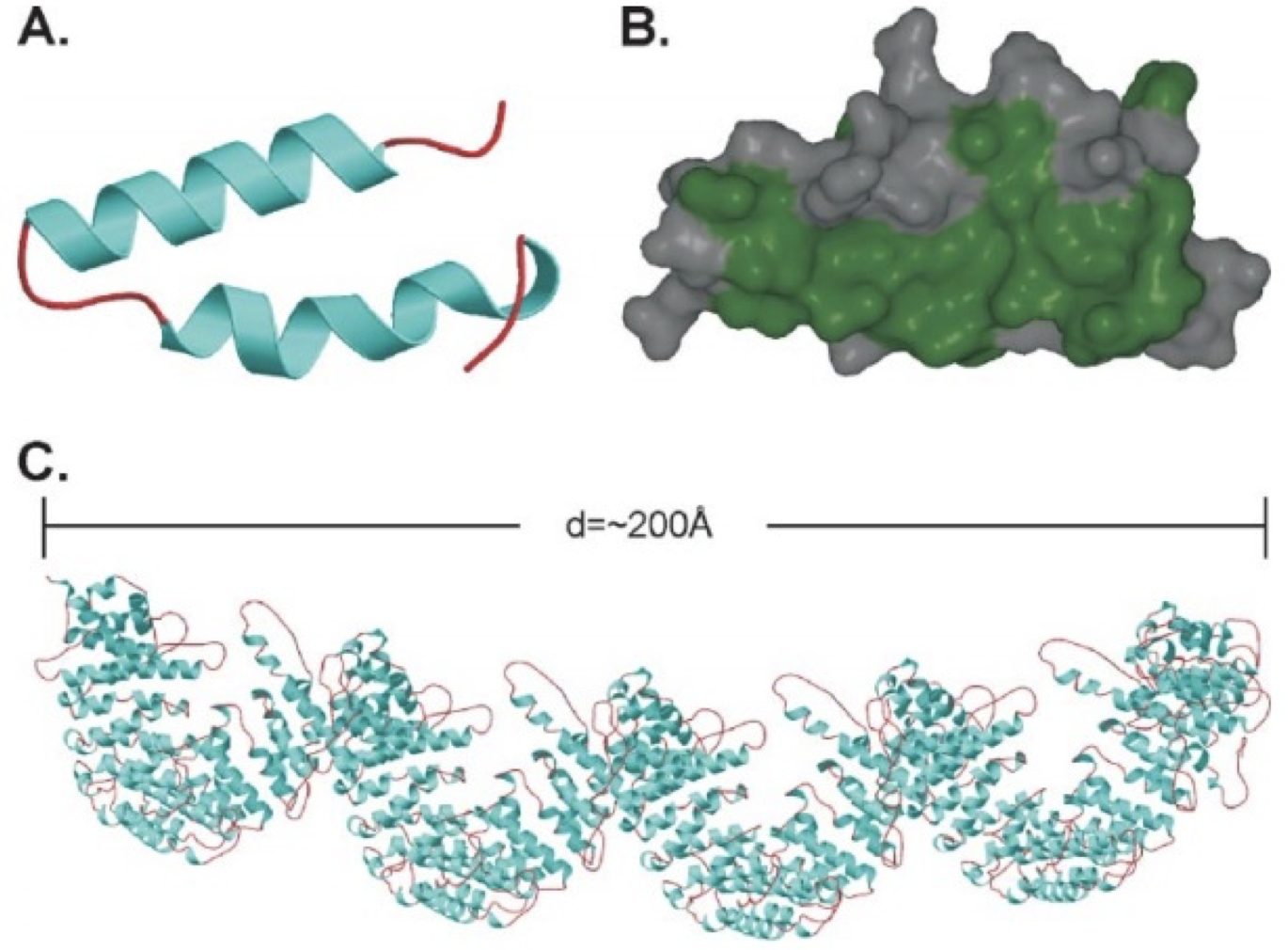
The IT15 (interesting transcript 15) gene located on chromosome 4p16.3 codes for the Huntingtin protein (Huntington's Disease Collaborative Research Project, 1993). Huntingtin itself is a large, 350kDa 3000 amino acid protein. It is mainly structured of large, 50 amino acid long repeats, termed HEAT repeats (from Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR kinase) (Andrade & Bork, 1995). These repeats are found in a number of different proteins and form hairpin helical structures. The repeat units then assemble into a superhelix with a continuous hydrophobic core seen in figure 2 (Li, Serpell, Carter, Ruiensztein, & Huntington, 2006).
Before continuing, an interesting point to note is that as the function of Htt is currently only theorised, there is no function-based assay available. This means studies that claim to have purified Htt cannot be sure that this is actually the material they have obtained.
The N terminus of Htt contains the crucial polyQ region. When this is expanded beyond the safe number of 36 repeats, the resulting conformational change causes multiple knock-on effects, including the formation of truncated protein (Ross & Tabrizi, 2011). To fully understand the effects of the mutant protein, knowledge of the role of wild type Huntingtin (wtHtt) is essential.
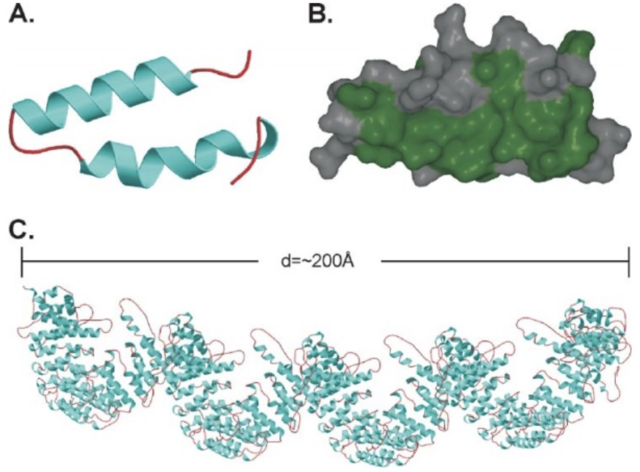 Figure 2. HEAT repeats. A shows a single helix turn helix HEAT repeat (helical hairpin). Both the helices (blue) and the coil regions (red) can vary in length. B shows the hydrophobic residues (green) of this structure. C shows a 2880-residue protein consisting of HEAT repeats, which can be assumed to be similar to the main structure of Htt. The superhelix structure is clearly visible. The study by Li et al (2006) that created this protein pointed out that as every red region in this protein was flexible, this showed why Htt was susceptible to proteolysis along its entire length. Figure from (Li, Serpell, Carter, Ruiensztein, & Huntington, 2006).
Figure 2. HEAT repeats. A shows a single helix turn helix HEAT repeat (helical hairpin). Both the helices (blue) and the coil regions (red) can vary in length. B shows the hydrophobic residues (green) of this structure. C shows a 2880-residue protein consisting of HEAT repeats, which can be assumed to be similar to the main structure of Htt. The superhelix structure is clearly visible. The study by Li et al (2006) that created this protein pointed out that as every red region in this protein was flexible, this showed why Htt was susceptible to proteolysis along its entire length. Figure from (Li, Serpell, Carter, Ruiensztein, & Huntington, 2006).
Much research has been conducted into the cellular function of Htt. Generally, when a new protein is discovered, it is sequenced to look for recognisable domains (e.g. ATP binding), which would provide some clue to function. However, when Htt was first analysed, it became clear that it had no recognisable domains (Landles & Bates, 2004). As well as this, the protein is expressed in many different subcellular compartments.
Research into the protein’s function has shown that it is both complex and important. A study by Rigamonti et al. (2000) found that wild type Huntingtin protected CNS neurons from apoptotic stimuli such as death receptor activation. Apoptotic cellular death is controlled by caspase proteins and wtHtt acted as a caspase-3 inhibitor in some mechanism upstream of its activation pathway. It was also shown that this protective effect required the portion of the protein between amino acid 63-548 to be present (Rigamonti, et al., 2000). Huntingtin is crucial for early embryonic development, as knock-out mouse models for their Huntingtin homolog protein (Hdh) show embryonic lethality within 8 days (Duyao, et al., 1995). This is attributed to increased apoptosis (Riener, Dragatsis, Zeitlin, & Goldowitz, 2003), linking to the protective role of Htt.
In hypomorphic Hdh mutations (protein reduced to around 1/3 of normal levels), early embryonic lethality is avoided but is replaced by severe developmental difficulties. These include defective neuron development and macroscopic malformations of the brain (White, et al., 1997). Interestingly, these characteristics are not shown in mice that express normal levels of the mutant form of Htt, indicating muHtt does not lose its neurogenic function. This confirms that HD is a gain of function mutation.
Due to the early embryonic death of Htt knockout mice, study of the long term effect of a lack of the protein is difficult. Another model organism that can be investigated is Drosophilia melangosta which also has an Htt homolog, known as dHtt (Li, Karlovich, Fish, Scott, & Myers, 1999). Unlike mouse models, dHtt knock out flies survive gestation and are born as normal. They also show a normal rate of development and no morphological differences but have reductions in mobility and fertility later in life (Zhang, Feany, Saraswati, Littleton, & Perrimon, 2009). The reason for the relatively normal development has been hypothesised to either be down to a compensatory mechanism, or defects that do not appear under normal conditions and would instead become apparent when the flies were stressed (Zheng & Joinnides, 2009).
Huntingtin has roles in maintaining the function of the cytoskeleton, which are reliant on interactions with HAP1 (Huntingtin-associated protein), which itself interacts with proteins such as kinesin and dynein (microtubule transporters, opposite directions) and dynactin (responsible for binding dynein and kinesin to the molecule requiring transport) (Caviston & Holzbaur, Huntingtin as an essential integrator of intracellular vesicle trafficking, 2009).
There are three possible models for the role of Htt in transport. It could be a switch, with phosphorylation of the protein acting to change transport direction along microtubules. Colin et al. (2008) showed serine phosphorylation at position 421 resulted in increased amounts of anterograde transport along cellular microtubules, whereas the unphosphorylated residue was associated with retrograde transport. The exact reason for this is unknown, but it is likely that the phosphorylated Htt protein has an increased affinity for acting as a scaffold protein between kinesin and dynactin (due to the direction kinesin travels in), whereas the dephosphorylated version enhances interaction between dynein and dynactin.
The second model of Htt in transport involves HAP40. When associated with Htt, HAP40 has been shown to affect whether vesicles are associated with actin filaments, used for short-range transport, or microtubules, used for long range transport (Pal, Severin, Lommer, Shevchenko, & Zerial, 2006). Htt/HAP40 complexes increase the association of vesicles with actin and as HAP40 is upregulated in HD (Pal, Severin, Lommer, Shevchenko, & Zerial, 2006), this could suggest a disease mechanism- defective long range cellular transport as a result of a greater than normal vesicle actin. Aberrant transcription of HAP40 may be why it is upregulated in HD.
The final potential model is that Htt only interacts with dynein and depletion of wtHtt results in defective and bi-directional dynein-bound vesicle transport, along with detachment of vesicles from transporters (Caviston, Ross, Antony, Tokito, & Holzbaur, 2007). This would result in cellular recycling pathways becoming compromised, potentially leading to a build-up of toxic material or depletion of crucial cellular components, again suggesting a role in the pathogenesis of HD.
Overall, there is abundant evidence that Htt is implicated in cellular transport. It is very likely that all three of the above models are correct and Htt is intimately involved in all stages of the transport process; forming a complex to bind vesicles to transporters, selecting the direction of travel and then detaching the vesicle at the target site. This complexity would explain why lowered levels of wtHtt have such a significant effect on the body.
Huntingtin has been shown in multiple studies to have direct interactions with transcription factors and transcription co-activators such as NFκB, p53 and CBP (Stefan, et al., 2000)(Takano & Gusella, 2002). As well as this, wtHtt interacts with the transcription activator Sp1, which itself is essential for recruitment of TFIID to the nucleus (Dunaw, et al., 2002). TFIID is a complex of proteins including TATA binding protein, used to bind to the TATA sequence present in the promoter sequences of certain eukaryotic genes. wtHtt binds to both Sp1 and TFII130, a component of TFIID, once again suggesting a role for Htt as a scaffold protein.
Other research has suggested that wtHtt is a transcription repressor. Htt binds to the N-CoR and Sin-3a receptor complex, used to prevent transcription of certain genes targeted by ligand activated nuclear receptors, such as retinoic acid receptor (Boutell, et al., 1999). It is clear Htt is involved in gene transcription, possibly promoting the synthesis of some genes and preventing synthesis of others to have an unknown combined effect.
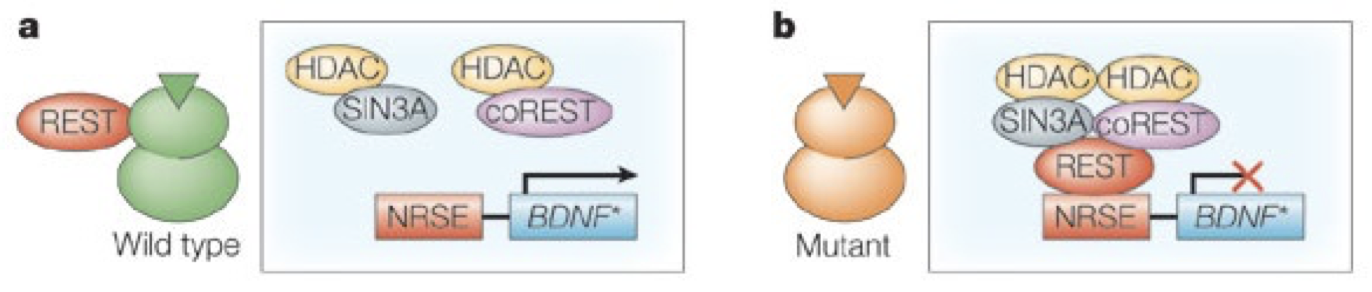
The most interesting role of Htt in transcription involves the production of a molecule called BDNF (brain derived neurotrophic factor). This acts on neurons to promote growth, survival and differentiation, forming new synapses (Huang & Reichardt, 2001). The BDNF gene is silenced by a molecule called REST/NRSF (RE1 silencing transcription factor/neuron restrictive silencer factor). REST/NRSF binds to the neuron restrictive silencing element (NRSE) in the BDNF promoter, recruiting a protein complex to inhibit synthesis (Fig. 3). Htt binds to this molecule, sequestering it (Zuccato, et al., 2003). Resultantly, levels of BDNF synthesis are increased and neuron development is stimulated.
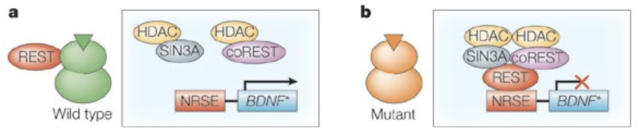 Figure 3. The REST/NRSF system. In the case of wtHtt (A), REST/NRSF is bound by the Htt molecule. This prevents it from binding to the NRSE, thereby preventing formation of the repressor complex, allowing synthesis of the BDNF gene. In the case of HD (B), muHtt does not effectively bind REST/NRSF, causing it to accumulate in the nucleus, repressing transcription of the BDNF gene. Figure adapted from (Cattaneo, Zuccato, & Tartari, 2005)
Figure 3. The REST/NRSF system. In the case of wtHtt (A), REST/NRSF is bound by the Htt molecule. This prevents it from binding to the NRSE, thereby preventing formation of the repressor complex, allowing synthesis of the BDNF gene. In the case of HD (B), muHtt does not effectively bind REST/NRSF, causing it to accumulate in the nucleus, repressing transcription of the BDNF gene. Figure adapted from (Cattaneo, Zuccato, & Tartari, 2005)
This suggests a crucial pathogenic role of muHtt- as there is such catastrophic neuron loss in disease sufferers and this molecule is responsible for healthy neuron survival, clearly there is some link. Heterozygous Hdh +/- knockout mice show reduced levels of BDNF mRNA (Zuccato, et al., 2003) and homozygous -/- Hdh knockout mice show a greatly reduced level of BDNF mRNA, along with progressive neurodegeneration and sterility (Dragatsis, Levine, & Zeitlin, 2000). Further to this, it has been shown that muHtt does not bind NRSF effectively (Zuccato, et al., 2003). This leads to the accumulation of NRSF in the nucleus, repressing BDNF synthesis. Therefore, both a lack of wtHtt and defective binding ability of muHtt are responsible for a loss of BDNF, a key factor in development of HD.Continued on Next Page »
Adams, J. (2003). The proteasome: structure, function, and role in the cell. Cancer Treatment Reviews, 29, 3-9.
Almqvist, E., Bloch, M., Brinkman, R., & Crauford, D. H. (1999). A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. American Journal of Human Genetics, 64, 1293-1304.
Andrade, M., & Bork, P. (1995). HEAT repeats in the Huntington's disease protein. Nature Genetics, 11, 115-116.
Arrasate, M., Mitra, S., Schweitzer, E., Segal, M., & Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431, 805-810.
Aziz, N., Swaab, D., Pjil, H., & Roos, R. (2007). Hypothalamic dysfunction and neuroendocrine and metabolic alterations in Huntington's disease: clinical consequences and therapeutic implications. Reviews in Neuroscience, 18, 223-251.
Aziz, N., van der Burg, J., Landwhermeyer, G., Brundin, P., Stijnen, T., & Roos, R. (2008). Weight loss in Huntington's disease increases with higher CAG repeat number. Neurology, 71, 1506-1513.
Beister, A., Kraus, P., Kuhn, W., Dose, M., Weindl, A., & Gerlach, M. (2004). The N-methyl-D-aspartate antagonist memantine retards progression of Huntington's disease. Journal of Neural Transmission. Supplementum, 68, 117-122.
Bennett, E., Bence, N. J., & Kopito, R. (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular Cell, 17, 351-365.
Biglan, K., Zhang, Y., Long, J., Geschwind, M., Kang, G., & Killoran, A. (2013). Refining the diagnosis of Huntington's disease: the PREDICT-HD study. Frontiers in Aging Neuroscience, 5, 12.
Boudreau, R., McBride, J., Martins, I., Shen, S., Xing, Y., & Carter, B. (2009). Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Molecular Therapy, 17, 1053-1063.
Boutell, J., Thomas, P., Neal, J., Weston, V., Duce, J., & Harper, P. (1999). Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Human Molecular Genetics, 8, 1647-1655.
Browne, S., Bowling, A., MacGarvey, U., Baik, M., Berger, S., & Muqit, M. (1997). Oxidative damage and metabolic dysfunction in Huntington's disease: selective vunerability of the basal ganglia. Annals of Neurology, 41, 646-653.
Carmichael, J., Chattelier, J., Woolfson, A., Milstein, C., Fersht, A., & Rubinsztein, D. (2000). Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proceedings of the National Academy of Science, 97, 9701-9705.
Cattaneo, E., Zuccato, C., & Tartari, M. (2005). Normal huntingtin function: an alternative approach to Huntington's disease. Nature Reviews Neuroscience, 6, 919-930.
Caviston, J., & Holzbaur, E. (2009). Huntingtin as an essential integrator of intracellular vesicle trafficking. Trends in Cell Biology, 19, 147-155.
Caviston, J., Ross, J., Antony, S., Tokito, M., & Holzbaur, E. (2007). Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proceedings of the National Academy of Science, 104, 10045-10050.
Chen, J., Ondo, W., Dashtipour, K., & Swope, D. (2012). Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clinical Therapeutics, 34, 1487-1504.
Choo, Y., Johnson, G., MacDonald, M., Detloff, P., & Lesort, M. (2004). Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Human Molecular Genetics , 13, 1407-1420.
Colin, E., Zana, D., Liot, G., Rangone, H., Borrell- Pages, M., & Li, X. (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. The EMBO Journal, 27, 2124-2134.
Cummings, C., Sun, Y., Opal, P., Antallfy, B., Mestril, R., & Orr, H. (2001). Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Human Molecular Genetics, 10, 1511-1518.
Davenport, C. (1911). Hereditary in Relation to Eugenics (1st ed.). New York: New York H. Holt.
Diaz-Hernandez, M., Hernandez, F., Martin-Aparicio, E., Gomez-Ramos, P., Moran, M., & Castano, J. (2003). Neuronal induction of the immunoproteasome in Huntington's disease. Journal of Neuroscience, 23, 11653-11661.
Diaz-Hernandez, M., Valera, A., Moran, M., Gomez-Ramos, P., Alvarez-Castelao, B., & Castano, J. (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. Journal of Neurochemistry, 98, 1585-1596.
DiFiglia, M., Sapp, E., Chase, K., Davies, S., Bates, G., & Vonsattel, J. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystophic neurites in brain. Science, 277, 1990-1993.
DiFiglia, M., Sena-Esteves, M., Chase, K., Sapp, E., Pfister, E., & Sass, M. (2007). Therpeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral defects. Proceedings of the National Academy of Sciences, 104, 17204-17209.
Dinq, Q., Lewis, J., Strum, K., Dimayuga, E., Bruce-Keller, A., & J, D. (2002). Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. Journal of Biological Chemistry, 277, 13935-13942.
Dragatsis, I., Levine, M., & Zeitlin, S. (2000). Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nature Genetics, 26, 300-306.
Dragileva, E., Hendricks, A., Teed, A., Gillis, T., Lopez, E., & Friedberg, E. (2009). Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neurobiology Disorders, 33, 37-47.
Dunaw, J., Jeong, H., Griffin, A., Kim, Y., Standaert, G., & Hersch, M. (2002). Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science, 296, 2238-2243.
Duyao, M., Auerbach, A., Ryan, A., Persichetti, F., Barnes, G., & McNeil, S. (1995). Inactivation of the mouse Huntington's disease gene homolog Hdh. Science, 269, 407-410.
Evers, M., Tran, H., Zalachoras, I., Meijer, O., den Dunnen, J., & van Ommen, G. (2014). Preventing formation of toxic N-terminal fragments through antisense oligonucleotide-mediated protein modification. Nucleic Acid Therapeutics, 24, 4-12.
Faber, P., Alter, J., Macdonald, M., & Hart, A. (1999). Polyglutamine mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proceedings of the National Academy of Science, 96, 179-184.
Fischer, U., Janssen, K., & Schulze-Osthoff, K. (2007). Does caspase inhibtion promote clonogenic tumor growth. Cell Cycle, 6, 3048-3053.
Frank, S. (2014). Treatment of Huntington's disease. Neurotherapeutics, 11, 153-160.
Frank, S., Ondo, W., Fahn, S., Hunter, C., Oakes, D., & Plumb, S. (2008). A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clinical Neuropharmacology, 31, 127-133.
Gines, S., Seong, I., Fossale, E., Ivanova, E., Trettel, F., & Gusella, J. (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Human Molecular Genetics, 12, 497-508.
Graham, R., Deng, Y., Slow, E., Haigh, B., Bissada, N., & Lu, G. (2006). Cleavage at the caspase 6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell, 125, 1179-1191.
Gu, M., Gash, M., Mann, V., Javoy-Aqid, F., Cooper, J., & Schapira, A. (1996). Mitochondrial defect in Huntington's disease caudate nucleus. Annals of Neurology, 39, 385-389.
Guo, J., Fisher, K., Darcey, R., Cryen, J., & O'Driscoll, C. (2010). Therapeutic targeting in the silent era: advances in non-viral siRNA delivery. Molecular BioSystems, 6, 1143-1161.
Guzhova, I., Lazarev, V., Kaznacheeva, A., Ippolitova, M., Muronetz, V., & Kinev, A. (2011). Novel mechanism of Hsp70 chaperone-mediated prevention of polyglutamine aggregates in a cellular model of Huntington's disease. Human Molecular Genetics, 20, 3953-3963.
Hansson, O., Nylandsted, J., Castilho, R., Leist, M., Jaattela, M., & Brundin, P. (2003). Overexpression of heat shock protein 70 in R6/2 Huntington’s disease mice has only modest effects on disease progression. Brain Research, 970, 47-57.
Harper, P. (1992). The epidemiology of Huntington's disease. Human Genetics, 89, 365-376.
HD iPSC Consortium. (2012). Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell, 11, 264-278.
Heemskerk, A., & Roos, R. (2012). Aspiration pneumonia and death in Huntington's disease. PLoS Currents, 1, RRN1293.
Hu, J., Matsui, M., Gagnon, K., Schwartz, J., Gabillet, S., & Arar, K. (2009). Inhibiting expression of mutant huntingtin and ataxin-3 by targetting expanded CAG repeat RNAs. Nature Biotechnology, 27, 478-484.
Huang, E., & Reichardt, L. (2001). Neurotrophins: Role in neuronal development and function. Annual Reviews in Neuroscience, 24, 677-736.
Huntington Study Group. (1996). Unified Huntington's Disease Rating Scale: Reliability and Consistency. Movement Disorders, 11, 136-142.
Huntington Study Group. (2006). Tetrabenazine as antichorea therapy in Huntington's disease. Neurology, 66, 366-372.
Huntington, G. (1872). On Chorea. Medical and Surgical Reporter of Philadelphia, 26, 317-321.
Huntington's Disease Collaborative Research Project. (1993). A novel gene containing a trinucelotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971-983.
Im, W., Ban, J., Lim, J., Lee, M., Lee, S., & Soo, K. (2013). Extracts of adipose derived stem cells slows progression in the R6/2 model of Huntington's disease. PLOS ONE, 8, e59438.
Imarisio, S., Carmichael, J., Korolchuk, V., Chen, C., Saiki, S., & Rose, C. (2008). Huntington's disease: from pathology and genetics to potential therapies. Biocemical Journal, 412, 191-209.
International Huntington Association. (1994). Guidelines for the molecular genetics predictive test in Huntington's disease. Neurology, 44, 1533-1536.
Jackson, G., Salecker, I., Dong, X., Yao, X., Arnheim, N., & Faber, P. (1998). Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron, 21, 633-642.
Jacobsen, J., Bawden, C., Rudiger, S., McLaughlan, C., Reid, S., & Waldvogel, H. (2010). An ovine transgenic Huntington’s disease model. Human Molecular Genetics, 19, 1873-1882.
Julien, C., Thompson, J., Wild, S., Yardumian, P., Snowden, J., & Turner, G. (2007). Psychiatric disorders in preclinical Huntington's disease. Journal of Neurology, Neurosurgery and Psychiatry, 78, 939-943.
Kornhuber, J., Weller, M., Schoppmeyer, K., & Riederer, P. (1994). Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. Journal of Neural Transmission: Supplementation, 43, 91-104.
Kovtun, I., Liu, Y., Bjoras, M., Klungland, A., Wilson, S., & McMurray, C. (2007). OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature, 447, 447-452.
Landles, C., & Bates, G. (2004). Huntingtin and the molecular pathogenesis of Huntington's disease. EMBO Reports, 5, 958-963.
Langbehn, D., Brinkman, R., Falush, D., Paulsen, J., & Hayden, M. (2004). A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clinical Genetics, 65, 266-267.
Lee, S., Cho, K., Jung, K., Im, W., Park, J., & Lim, H. (2009). Slowed progression in models of Huntington's disease by adipose stem cell transplantation. Annals of Neurology, 66, 671-681.
Li, H., Li, S., Yu, Z., Shelbourne, P., & Li, X. (2001). Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. Journal of Neuroscience, 21, 8473-8481.
Li, J., Popovic, N., & Brundin, P. (2005). The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. Journal of the American Society for Experimental Neurotherapeutics, 2, 447-464.
Li, S., & Li, X. (2006). Multiple pathways contribute to the pathogenesis of Huntington's disease. Molecular Neurodegeneration, 1, 19.
Li, W., Serpell, L., Carter, W., Ruiensztein, D., & Huntington, J. (2006). Expression and characterisation of full length human huntingtin, an elongated HEAT repeat protein. Journal of Biological Chemistry, 281, 15916-15922.
Li, Z., Karlovich, C., Fish, M., Scott, M., & Myers, R. (1999). A putative Drosophila homolog of the Huntington’s disease gene. Human Molecular Genetics, 8, 1807-1815.
Lin, Y., & Wilson, J. (2007). Trascription induced CAG repeat contraction in human cells is mediated in part by transcription coupled nucleotide excision repair. Molecular & Cellular Biology, 27, 6209-6217.
Lin, Y., Chern, Y., Shen, C., Wen, H., Chagn, Y., & Li, H. (2011). Human mesenchymal stem cells prolong survival and ameliorate motor deficit through trophic support in Huntington's disease mouse models. PLoS One, 6, e22924.
Lombardi, M., Jaspers, L., Spronkmans, C., Gellera, C., Taroni, F., & Di Maria, E. (2009). A majority of Huntington's disease patients may be treatable by individualized allele-specific RNA interference. Experimental Neurology, 217, 312-319.
Lopatina, T., Kalinina, N., Karagyuar, M., Stambolsky, D., Rubina, K., & Revischin, A. (2011). Adipose-derived stem cells stimulate regeneration of peripheral nerves: BDNF secreted by these cells promotes nerve healing and axon growth de novo. PLOS ONE, 6, e17899.
Lovestone, S., Hodgson, S., Sham, P., Differ, A., & Levy, R. (1996). Familial psychiatric presentation of Huntington's disease. Journal of Medical Genetics, 33, 128-131.
Lucetti, C., Gambacinni, G., Bernardini, S., Dell'Agnello, G., Petrozzi, L., & Rossi, G. (2002). Amantadine in Huntington’s disease: open-label video-blinded study. Neurological Sciences, 23, 83-84.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., & Hetherington, C. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell, 87, 493-506.
McBride, H., Neuspiel, M., & Wasiak, S. (2006). Mitochondria: more than just a powerhouse. Current Biology, 16, 551-560.
McGeer, E., & McGeer, P. (1976). Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature, 263, 517-519.
McGill, J., & Beal, M. (2006). PGC-1alpha, a new therapeutic target in Huntington's disease. Cell, 127, 465-468.
McMurray, C. (2010). Mechanisms of trinucleotide repeat instability during human development. Nature Reviews Genetics, 11, 786-799.
Milakovic, T., & Johnson, G. (2005). Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant Huntingtin. Journal of Biological Chemistry, 280, 30773-30782.
Millipore. (2014). Anti-Huntingtin protein antibody, clone mEM48. Retrieved 03 06, 2014, from http://www.millipore.com/catalogue/item/mab5374
Morton, J., Wood, N., Hastings, M., Hurelbrink, C., Barker, R., & Maywood, E. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. Journal of Neuroscience, 25, 157-163.
Niclis, J., Trounson, A., Dottori, M., Ellisdon, A., Bottomley, S., & Verlinksy, Y. (2009). Human embryonic stem cell models of Huntington's disease. Reproductive Biomedicine Online, 19, 106-113.
Ona, V., Li, M., Vonsattel, J., Andrews, L., Khan, S., & Chung, W. (1999). Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature, 399, 263-267.
O'Suilleabhain, P., & Dewey, R. (2003). A randomised trial of amantadine in Huntington's disease. Archives of Neurology, 60, 996-998.
Pal, E., Severin, F., Lommer, B., Shevchenko, A., & Zerial, M. (2006). Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. Journal of Cell Biology, 72, 605-618.
Panov, A., Lund, S., & Greenamyre, J. (2005). Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington's disease individuals. Molecular & Cellular Biochemistry, 269, 143-152.
Perluigi, M., Poon, H., Maragos, W., Pierce, W., Klein, J., & Calabrese, V. (2005). Proteomic analysis of protein expression and oxidative modification in R6/2 trangenic mice. Molecular & Cellular Proteomics, 4, 1849-1861.
Perrin, V., Regulier, E., Abbas-Terki, T., Hassig, R., Brouillet, E., & Aebischer, P. (2007). Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Molecular Therapy, 15, 903-911.
Pfister, E., & Zamore, P. (2009). Huntington's disease: silencing a brutal killer. Experimental Neurology, 220, 226-229.
Polidori, M., Mecocci, P., Browne, S., Senin, U., & Beal, M. (1999). Oxidative damage to mitochondrial DNA in Huntington's disease parietal cortex. Neuroscience Letters, 272, 53-56.
Pouladi, M., Morton, J., & Hayden, M. (2013). Choosing an animal model for the study of Huntington's disease. Nature Reviews Neuroscience, 14, 708-721.
Ravikumar, B., Vacher, C., Berger, Z., Davies, J., Lou, S., & Oroz, L. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington's disease. Nature Genetics, 36, 585-595.
Reddy, P., Charles, V., Williams, M., Miller, G., Whetsell, W., & Tagle, D. (1999). Transgenic mice expressing mutated full-length HD cDNA: a paradigm for locomotor changes and selective neuronal loss in Huntington's disease. Philosophical Transactions of the Royal Society of London Biological Sciences, 354, 1035-1045.
Ridley, R., Frith, C., Crow, T., & Conneally, P. (1988). Anticipation in Huntington's disease is inherited through the male line but may originate in the female. Journal of Medical Genetics, 25, 589-595.
Riener, A., Dragatsis, I., Zeitlin, S., & Goldowitz, D. (2003). Wild-type Huntingtin plays a role in brain development and neuronal survival. Molecular Neurobiology, 28, 259-275.
Rigamonti, D., Bauer, J., De-Fraja, C., Conti, L., Sipione, S., & Sciorati, C. (2000). Wild-type Huntingtin protects from apoptosis upstream of caspase-3. Journal of Neuroscience, 20, 3705-3713.
Roos, R. (2010). Huntington's disease: a clinical review. Orphanet Journal of Rare Diseases, 5, 40.
Rosenblatt, A. (2007). Understanding the psychiatric prodome of Huntington's disease. Journal of Neurology, Neurosurgery and Psychiatry, 78, 913.
Ross, C., & Tabrizi, S. (2011). Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurology, 10, 83-98.
Rossignol, J., Boyer, C., Leveque, X., Finq, K., Thinard, R., & Blanchard, F. (2011). Mesenchymal stem cell transplantation and DMEM administration in a 3NP rat model of Huntington's disease: morphological and behavioural outcomes. Behavioural Brain Research, 217, 369-378.
Rossignol, J., Fink, K., Davies, K., Clerc, S., Crane, A., & Matchynski, J. (2014). Transplant of adult mesenchymal and neural stem cells provide neuroprotection and behavioural sparing in a transgenic rat model of Huntington's disease. Stem Cells, 32, 500-509.
Ryu, J., Kim, J., Cho, S., Hatori, K., Nagai, A., & Choi, H. (2004). Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiology of Disease, 16, 68-77.
Saxton, W., & Hollenbeck, P. (2012). The axonal transport of mitochondria. Journal of Cell Biology, 125, 2095-2104.
Schwarz, D., Hutvagner, G., Du, T., Xu, Z., Aronin, N., & Zamore, P. (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell, 115, 199-208.
Sieradzan, K., Mechan, A., Jones, L., Wanker, E., Nukina, N., & Mann, D. (1999). Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Experimental Neurology, 156, 92-99.
Slow, E., Graham, R., Osmand, A., Devon, R., Lu, G., & Deng, G. (2005). Absence of behavioural abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proceedings of the National Academy of Science, 102, 11402-11407.
Stefan, J., Kazantsev, A., Spasiv-Boscovic, O., Greenwald, M., Zhu, Y., & Gohler, H. (2000). The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proceedings of the National Academy of Science, 97, 6763-6768.
Strachan, T., & Read, A. (2011). Human Molecular Genetics. In E. Owen (Ed.). Garland Science.
Summerton, J. (2007). Morpholino, siRNA and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Current Topics in Medicinal Chemistry, 7, 651-660.
Tabrizi, S., Reilmann, R., Roos, R., Durr, A., Leavitt, B., & Owen, G. (2012). Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurology, 11, 42-53.
Takano, H., & Gusella, J. (2002). The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. Biomed Central Neuroscience, 3, 15.
Tian, J., Yan, Y., Zhou, R., Lou, H., Rong, Y., & Zhang, B. (2014). Soluble N-terminal fragment of mutant huntingtin protein impairs mitochondrial axonal transport in cultured hippocampal neurons. Neuroscience Bulletin, 30, 74-80.
Tomar, R., Matta, H., & Chaudhary, P. (2003). Use of adeno-associated viral vector for delivery of small interfering RNA. Oncogene, 22, 5712-5715.
Tyagi, S., Tyagi, L., Shekhar, R., Singh, M., & Kori, M. (2010). Symptomatic treatment and management of Huntington's disease: an overview. Global Journal of Pharmacology, 4, 6-12.
Vacher, C., Garcia-Oroz, L., & Rubinsztein, D. (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Human Molecular Genetics, 14, 3425-3433.
van Wamelen, D., Aziz, N., Anink, J., Steenhoven, R., Angeloni, D., & Fraschini, F. (2013). Suprachiasmatic nucleus neuropeptide expression in patients with Huntington's disease. Sleep, 36, 117-125.
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., & Goldberg, A. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Molecular Cell, 14, 95-104.
Violante, V., Luongo, A., Pepe, I., Annunziata, S., & Gentile, V. (2001). Transglutaminase-dependent formation of protein aggregates as possible biochemical mechanism for polyglutamine diseases. Brain Research Bulletin, 56, 169-172.
Walker, F. (2007). Huntington's disease. The Lancet, 369, 218-228.
Warby, S., Montpetit, A., Hayden, A., Carroll, J., Butland, S., & Visscher, H. (2009). CAG expansion in the Huntington disease gene is associated with a specific and targettable predisposing haplogroup. The American Journal of Human Genetics, 84, 351-366.
White, J., Auerback, W., Duyao, M., Vonsattel, J., Gusella, J., & Joyner, A. (1997). Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nature Genetics, 17, 404-410.
Whitehead, K., Langer, R., & Anderson, D. (2009). Knocking down barriers: advances in siRNA delivery. Nature Reviews Drug Discovery, 129-138.
Wilkins, A., Kemp, K., Ginty, M., Hares, K., Mallam, E., & Scolding, N. (2009). Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Research, 3, 63-70.
Yamamoto, A., Lucas, J., & Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell, 101, 57-66.
Yang, D., Wang, C., Zhao, B., Li, W., Ouyang, Z., & Liu, Z. (2010). Expression of Huntington's disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Human Molecular Genetics, 19, 3983-3994.
Yang, S., Cheng, P., Banta, H., Piotrowska-Nitsche, K., Yang, J., & Cheng, E. (2008). Towards a transgenic model of Huntington's disease in a non-human primate. Nature, 453, 921-924.
Yang, W., Dunlap, J., Andrews, R., & Wetzel, R. (2002). Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Human Molecular Genetics, 11, 2905-2917.
Yu, J., Vodyanik, M., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J., & Tian, S. (2007). Induced pluripotent stem cell lines generated from human somatic cells. Science, 318, 1917-1920.
Zhang, S., Feany, M., Saraswati, S., Littleton, J., & Perrimon, N. (2009). Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington's disease model. Disease Models & Mechanisms, 2, 247-266.
Zhang, Y., Li, M., Drozda, M., Chen, M., Ren, S., & Mejia-Sanchez, R. (2003). Depletion of wild-type huntingtin in mouse models of neurological diseases. Journal of Neurochemistry, 87, 101-106.
Zheng, Q., & Joinnides, M. (2009). Hunting for the function of Huntingtin. Disease Models & Mechanisms, 2, 199-200.
Zhou, H., Cao, F., Wang, Z., Yu, Z., Nguyen, H., & Evans, J. (2003). Huntingtin forms toxic NH2 terminal fragment complexes that are promoted by the age-dependant decrease in proteasome activity. Journal of Cell Biology, 163, 109-118.
Zoghby, H., & Orr, H. (2000). Glutamine repeats and neurodegeneration. Annual Reviews in Neuroscience, 23, 217-247.
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., & Conti, L. (2003). Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature Genetics, 35, 76-83.